BNConverter can learn Bayesian network parameters from data for arbitrarily structured networks; these can be evaluated immediately or with BNTester to predict functional relationships. This behavior can be modified in a number of ways for evaluation, including different learning algorithms, training/test splits, and randomizations.
Overview
Unlike many Sleipnir Bayesian network tools, most of which are specialized for naive Bayesian classifiers, BNConverter can learn parameters for and evaluate arbitrarily structured networks. These can include unobserved (hidden) nodes, multiple parent/child relationships, and so forth.
Bayesian integration generally entails assigning one Bayesian network node to each available biological dataset. Groups of related datasets (e.g. all physical binding datasets) can be collected under a single unobserved "parent" node, and the network is capped by a single Functional Relationship (FR) node representing whether a particular observation (e.g. gene pair) is functionally related. This process is detailed in Troyanskaya et al 2003.
Given such a network (usually as a SMILE DSL or XDSL file), a collection of discretized biological datasets (usually Sleipnir::CDat s stored as DAT or DAB files with associated QUANT files), and a functional gold standard (usually a Sleipnir::CDat), BNConverter will learn the conditional probabilities associated with each dataset and value. Conversely, given a Bayesian network and biological datasets without a gold standard, BNConverter will evaluate the network to infer probabilities of functional relationship based on all available data.
For example, consider the Bayesian network used in Myers et al 2005, which we'll assume we've saved as biopixie.xdsl:
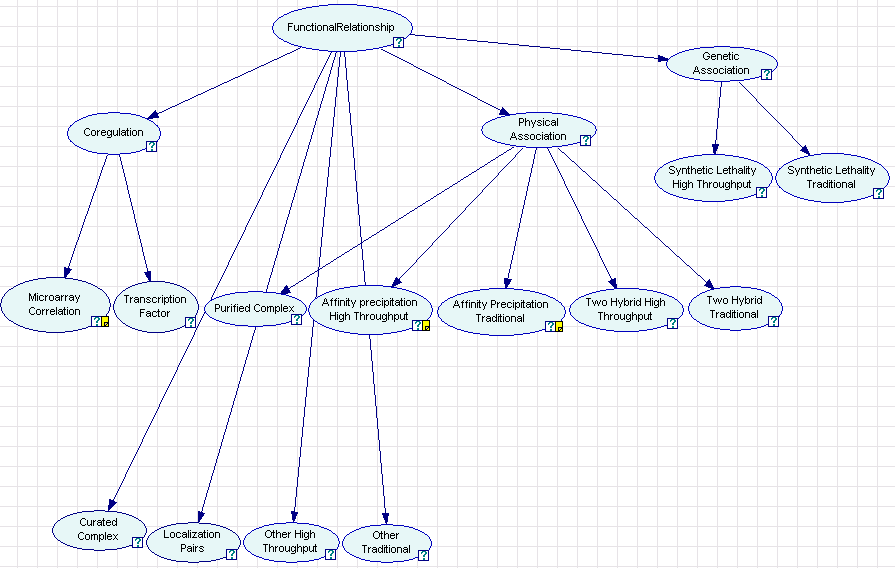
The name of each node is displayed, and SMILE associated an ID with each name; this might be FR for FunctionalRelationship, COREGULATION for Coregulation, MICROARRAY for Microarray Correlation, and so forth. Each leaf node corresponds to a single dataset, and each non-leaf node is an unobserved (hidden) and has no associated dataset. To learn parameters for this network, we should assemble a directory of data files:
MICROARRAY.dab MICROARRAY.quant TF.dab TF.quant CUR_COMPLEX.dab CUR_COMPLEX.quant ... SYNL_TRAD.dab SYNL_TRAD.quant
Each data file is a Sleipnir::CDat, either a DAT or a DAB, containing experimental results. Each QUANT file describes how to discretize that data for use with the Bayesian network (the number of bins in the QUANT must be the same as the number of values taken by the corresponding node in the Bayesian network). Once we've placed all of these files in a directory (e.g. ./data/) and assembled a functional gold standard (e.g. ANSWERS.dab, possibly constucted by Answerer), we can learn the network's conditional probabilities using Expectation Maximization:
BNConverter -d ./data/ -i biopixie.xdsl -o learned.xdsl -w ANSWERS.dab
This produces a "learned" network with probabilities that model (as accurately as possible) the relationship between the given data and functional gold standard. This model can be used to predict functional relationships between new genes not in the standard (but with experimental data) using BNTester or the -e and -E evaluation arguments, e.g.
BNConverter -d ./data/ -i biopixie.xdsl -o learned.xdsl -w ANSWERS.dab -t 0.25
-e heldout_gene_predictions.dab
The heldout_gene_predictions.dab file now containins a Sleipnir::CDat in which each pairwise score represents a probability of functional relationship, and it can be mined with tools such as Dat2Dab or Dat2Graph.
Usage
Basic Usage
BNConverter -d <data_dir> -i <network.xdsl> -o <learned.xdsl> -w <answers.dab> -t <frac>
-e <test_predictions.dab> -E <train_predictions.dab>
Saves learned parameters for the network network.xdsl in the new network learned.xdsl, based on the data in data_dir (containing files with names corresponding to the network node IDs) and the functional gold standard in answers.dab. Hold frac fraction of the gene pairs out of training; store predicted probabilities of functional relationship for these pairs in test_predictions.dab and the remaining inferred probabilities in train_predictions.dab.
Detailed Usage
package "BNConverter"
version "1.0"
purpose "Bayes net training and testing"
defgroup "Data" yes
groupoption "datadir" d "Data directory"
string typestr="directory" group="Data"
groupoption "dataset" D "Dataset DAD file"
string typestr="filename" group="Data"
section "Main"
option "input" i "Input (X)DSL file"
string typestr="filename" yes
option "output" o "Output (X)DSL or DAT/DAB file"
string typestr="filename" yes
option "answers" w "Answer DAT/DAB file"
string typestr="filename"
section "Learning/Evaluation"
option "genes" g "Gene inclusion file"
string typestr="filename"
option "genex" G "Gene exclusion file"
string typestr="filename"
option "genet" c "Term inclusion file"
string typestr="filename"
option "randomize" a "Randomize CPTs before training"
flag off
option "murder" m "Kill the specified CPT before evaluation"
int
option "test" t "Test fraction"
double default="0"
option "eval_train" E "Training evaluation results"
string typestr="filename"
option "eval_test" e "Test evaluation results"
string typestr="filename"
section "Network Features"
option "default" b "Bayes net containing defaults for cases with missing data"
string typestr="filename"
option "zero" z "Zero missing values"
flag off
option "elr" l "Use ELR algorithm for learning"
flag off
option "pnl" p "Use PNL library"
flag off
option "function" f "Use function-fitting networks"
flag off
section "Optional"
option "group" u "Group identical inputs"
flag on
option "iterations" s "EM iterations"
int default="20"
option "checkpoint" k "Checkpoint outputs after each iteration"
flag off
option "random" r "Seed random generator"
int default="0"
option "verbosity" v "Message verbosity"
int default="5"
| Flag | Default | Type | Description |
|---|---|---|---|
| -d | None | Directory | Directory containing data files. Must be DAB, DAT, DAS, or PCL files with associated QUANT files (unless a continuous network is being learned) and names corresponding to the network node IDs. |
| -D | None | DAD file | DAD file containing data and/or answers for Bayesian learning or evaluation. Generally constructed using Dab2Dad. |
| -i | None | (X)DSL file | File from which Bayesian network structure and/or parameters are determined. During learning, only the structure is used; during evaluation, both structure and parameters are used. |
| -o | None | (X)DSL or DAT/DAB file | During learning, (X)DSL file into which a copy of the Bayesian network with learned parameters is stored. During evaluation, DAT or DAB file in which predicted probabilities of functional relationship are saved. |
| -w | None | DAT/DAB file | Functional gold standard for learning. Should consist of gene pairs with scores of 0 (unrelated), 1 (related), or missing (NaN). |
| -b | None | (X)DSL file | If present during learning, parameters from the given (X)DSL file are used instead of learned parameters for probability tables with too few examples. For details, see Sleipnir::CBayesNetSmile::SetDefault. |
| -g | None | Text gene list | If given, use only gene pairs for which both genes are in the list. For details, see Sleipnir::CDat::FilterGenes. |
| -G | None | Text gene list | If given, use only gene pairs for which neither gene is in the list. For details, see Sleipnir::CDat::FilterGenes. |
| -c | None | Text gene list | If given, use only gene pairs passing a "term" filter against the list. For details, see Sleipnir::CDat::FilterGenes. |
| -a | off | Flag | If on, randomize all parameters before learning or evaluation. |
| -m | None | Integer | If given, randomize the parameters of the network node at the given index. |
| -t | 0 | Double | Fraction of available gene pairs to randomly withhold from training and use for evaluation. |
| -E | None | DAT/DAB file | If given, save predicted probabilities of functional relationship for the training gene pairs in the requested file. |
| -e | None | DAT/DAB file | If given, save predicted probabilities of functional relationship for the test gene pairs in the requested file. |
| -z | off | Flag | If on, assume that all missing gene pairs in all datasets have a value of 0 (i.e. the first bin). |
| -l | off | Flag | If on, use the Extended Logistic Regression (ELR) algorithm for learning (due to Greiner and Zhou 2005) in place of EM. This will learn a discriminative model, whereas EM will learn a generative one. |
| -p | off | Flag | If on, use Intel's PNL library for Bayesian network manipulation rather than SMILE. Note that Sleipnir must be compiled with PNL support for this to function correctly! |
| -f | off | Flag | If on, assume the given (X)DSL file represents a custom function-fitting Bayesian network. For details, see Sleipnir::CBayesNetFN. |
| -u | on | Flag | If on, group identical examples into one heavily weighted example. This greatly improves efficiency, and there's essentially never a reason to deactivate it. |
