BNEvaluator will perform Bayesian inference on a given network by reading feature (node) values directly from a PCL file. Rather than accessing a collection of biological datasets stored as separate files, BNEvaluator reads examples from rows of a PCL file, assumes each experimental column is a feature (node), discretizes the data in each cell with an associated QUANT file, and infers probabilities for each possible value of a single class node (usually binary).
Overview
BNEvaluator can be used to perform Bayesian inference on arbitrary Bayes networks using data read directly from a PCL File (see Sleipnir::CPCL) Unlike most of Sleipnir's Bayesian analysis tools, which assume each gene pair is an example and each dataset a feature, BNEvaluator is agnostic to the form and semantics of the PCL's data: each row is an example, each experimental column a feature, and values in the cells are discretized using a single associated QUANT file (e.g. data.quant for a data file data.pcl ).
For example, suppose we have a data file data.pcl containing:
ID DATA1 DATA2 DATA3 THING1 1.1 2.2 3.3 THING2 2.1 3.2 1.3 THING3 3.0 2.0 1.0
In the same directory, we have a QUANT file containing:
1.5 2.5 3.5 4.5
Finally, we have a Bayesian network network.xdsl with reasonable parameters and the structure:
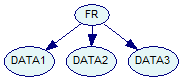
Each of the three data nodes can take four values (to agree with our QUANT file), and the class node FR is binary.
We now run (noting that our data.pcl has zero skip columns):
BNEvaluator -i network.xdsl -d data.pcl -s 0 -o results.pcl
This will evaluate the probability of the FR class node's values using the three examples [0, 1, 2], [1, 2, 0], and [2, 1, 0] for the data nodes and produce a results.pcl file that resembles:
ID FR0 FR1 THING1 0.3 0.7 THING2 0.9 0.1 THING3 0.4 0.6
Usage
Basic Usage
BNEvaluator -i <network.xdsl> -d <data.pcl> -o <results.pcl>
Saves inferred probabilities for the class (FR) node from the network network.xdsl in the output results.pcl file, based on the data (columns) for each example (row) in data.pcl.
Detailed Usage
package "BNEvaluator"
version "1.0"
purpose "Per-gene whole Bayes net evaluation"
section "Main"
option "input" i "Input (X)DSL file"
string typestr="filename" yes
option "data" d "Input PCL file"
string typestr="filename" yes
section "Miscellaneous"
option "output" o "Output PCL file"
string typestr="filename"
option "skip" s "Columns to skip in input PCL"
int default="2"
section "Learning/Evaluation"
option "genes" g "Gene inclusion file"
string typestr="filename"
option "genex" G "Gene exclusion file"
string typestr="filename"
option "zero" z "Zero missing values"
flag off
section "Network Features"
option "pnl" p "Use PNL library"
flag off
option "function" f "Use function-fitting networks"
flag off
section "Optional"
option "algorithm" a "Bayesian inference algorithm"
int default="0"
option "group" u "Group identical inputs"
flag on
option "verbosity" v "Message verbosity"
int default="5"
| Flag | Default | Type | Description |
|---|---|---|---|
| -i | None | (X)DSL file | File from which Bayesian network structure and parameters are determined. Columns of the input PCL are mapped by name to node IDs in the given network. |
| -d | None | PCL text file | PCL file containing examples (rows) from which data features (columns) are read and quantized for use during evaluation of the given Bayes net. For a non-continuous Bayes net, must have an associated QUANT file of the same name (e.g. data.quant in the same location as a data file data.pcl). |
| -o | stdout | PCL text file | PCL file in which inferred class probabilities are saved. Each row is an example from the input file, each column a possible value of the class node (zero indexed). |
| -s | 2 | Integer | Number of columns to skip between the initial ID column and the first experimental (data) column in the input PCL. |
| -g | None | Text gene list | If given, use only genes in the list. |
| -G | None | Text gene list | If given, use only gene not in the list. |
| -z | off | Flag | If on, assume that all missing gene pairs in all datasets have a value of 0 (i.e. the first bin). |
| -p | off | Flag | If on, use Intel's PNL library for Bayesian network manipulation rather than SMILE. Note that Sleipnir must be compiled with PNL support for this to function correctly! |
| -f | off | Flag | If on, assume the given (X)DSL file represents a custom function-fitting Bayesian network. For details, see Sleipnir::CBayesNetFN. |
| -a | 0 | Integer | ID of Bayesian inference algorithm to use (passed directly to SMILE). The default is almost always the most efficient and accurate option. |
| -u | on | Flag | If on, group identical examples into one heavily weighted example. This greatly improves efficiency, and there's essentially never a reason to deactivate it. |
