Prepares prerequisite files that are necessary for the efficient integrations of coexpressions in SeekMiner and SeekServer. Some of the file preparation tasks that SeekPrep performs are: preparing gene-presence file, calculating gene average correlation, calculating gene expression variances for each dataset.
Usage
Basic Usage
Prepare Gene Average File (GAVG)
SeekPrep -i <gene_map> -d -B <dab_file> -a -D <output_dir>
Calculates the average z-score for each gene in a given DAB matrix and stores the results as a vector of floats in the GAVG file. The index of a gene in the vector is determined by gene_map.
Prepare Gene Presence File (GPRES)
SeekPrep -i <gene_map> -d -B <dab_file> -p -D <output_dir>
Stores the gene presence vector for a given DAB matrix, where each value is either 1 if the gene is present, or 0 if the gene is absent in the dataset.
Prepare Dataset Sinfo file (SINFO)
SeekPrep -i <gene_map> -e -V <pclbin_file> -s -D <output_dir>
Calculates the average Fisher's transformed correlation between all gene pairs in an input dataset. The input dataset needs to be a binary PCL file with the extension BIN (generated by PCL2Bin).
Prepare Dataset Gene Expression Variance file (GEXPVAR)
SeekPrep -i <gene_map> -e -V <pclbin_file> -v -D <output_dir>
Calculates the gene expression variance for each gene in an input dataset.
Prepare Platform average z-scores and their standard deviation (GPLAT)
SeekPrep -i <gene_map> -f -P -b <db_file_list> -I <prep_dir> -A <dset_platform_map> -Q <quant>
Calculates the platform-wide average of z-scores ( 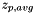 ) using the following algorithm:
) using the following algorithm:
For each dataset 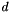 :
:
For each gene 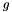 in the genome
in the genome 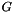 :
:
Compute 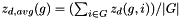
For each gene 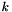 in the genome :
in the genome :
Compute 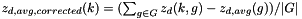
For each platform 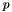 and its set of dataset
and its set of dataset 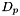 :
:
For each gene in the genome :
Compute 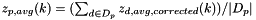
The prep_dir contains the GPRES and GAVG files for all datasets defined in dset_platform_map. (Users should generate these files with SeekPrep first.)
The dset_platform_map is a tab-delimited file that looks something like:
GSE15913.GPL570.pcl GPL570 GSE16122.GPL2005.pcl GPL2005 GSE16797.GPL570.pcl GPL570 GSE16836.GPL570.pcl GPL570 GSE17351.GPL570.pcl GPL570 GSE17537.GPL570.pcl GPL570
where the 1st column is the dataset name and the 2nd column is the corresponding platform.
The quant file is a space-delimited file that specifies how the z-scores are binned:
-5.00 -4.96 -4.92 -4.88 -4.84 -4.80 -4.76 -4.72 -4.68 -4.64 -4.60 -4.56 -4.52 ...
The db_file_list file is a list of file paths to the entire DB collections:
/x/y/z/00000001.db /x/y/z/00000002.db /x/y/z/00000003.db /x/y/z/00000004.db /x/y/z/00000005.db ...
Detailed Usage
package "SeekPrep"
version "1.0"
purpose "Preprocess datasets for Seek"
section "Mode"
option "dab" d "DAB mode, suitable for dataset wide gene average and stdev calculation"
flag off
option "pclbin" e "PCL BIN mode, suitable for dataset gene variance calculation"
flag off
option "db" f "DB mode, suitable for platform wide gene average and stdev calculation"
flag off
option "dabset" g "DAB set mode, sums a set of sparse rank-normalized (or subtract-z-normalized) DAB files, with weights or no weights"
flag off
option "combined_dab" h "Combined DAB mode, divides a summed DAB file by total pair counts or dataset weights, generates a new normalized DAB file"
flag off
section "Combined DAB mode"
option "dab_dir2" H "Directory containing the summed DAB file"
string typestr="directory" default="NA"
option "dab_basename" J "Summed DAB basename (ie without extension)"
string typestr="filename" default="NA"
section "DAB set mode (also see Misc options)"
option "dab_dir" G "Directory containing the DAB files"
string typestr="directory" default="NA"
option "dablist" L "List of DAB files"
string typestr="filename" default="NA"
option "out_dab" O "Output DAB file basename (ie without extension)"
string typestr="filename" default="NA"
option "dataset_w" W "Dataset weights (optional)"
string typestr="filename" default="NA"
section "DAB mode"
option "gavg" a "Generates gene average file"
flag off
option "gpres" p "Generates gene presence file"
flag off
option "dabinput" B "DAB dataset file"
string typestr="filename"
option "top_avg_percent" C "For gene average, top X percent of the values to take average (0 - 1.0)"
float default="1.0"
option "norm" F "Normalize matrix then sparsify it (needs --norm_mode)"
flag off
option "view" X "View distribution of values in the matrix"
flag off
section "PCL mode"
option "pclinput" V "PCL BIN file"
string typestr="filename"
option "gexpvarmean" v "Generates gene expression variance and mean files (.gexpvar, .gexpmean)"
flag off
option "sinfo" s "Generates sinfo file (dataset z score mean and stdev)"
flag off
section "DB mode"
option "gplat" P "Generates platform wide gene average and stdev file"
flag off
option "dblist" b "The DB file list (incl. file path)"
string typestr="filename"
option "dir_prep_in" I "The prep directory containing the .gavg and .gpres files"
string typestr="directory"
option "dset" A "The dataset platform mapping file"
string typestr="filename"
option "useNibble" N "If the DB is nibble type"
flag off
option "quant" Q "Quant file"
string typestr="filename"
section "Misc"
option "default_type" T "Default gene index type (choose unsigned short for genes, or unsigned int (32-bit) for transcripts) (required for DAB set mode and if --norm is enabled in DAB mode) (0 - unsigned int, 1 - unsigned short)"
int default="-1"
option "norm_mode" n "Normalization method: rank - rank-normalize matrix, subtract_z - subtract-z-normalize matrix (required for DAB set mode and if --norm is enabled, topological_overlap - TO measure from Ravasz et al)"
values="rank","subtract_z","topological_overlap","NA" default="NA"
option "logit" l "For --gavg and --gplat, whether to take logit of the value first (useful if edge value is probability)"
flag off
option "max_rank" M "Maximum rank value (for --norm_mode=rank)"
int default="-1"
option "rbp_p" R "RBP p parameter (for --norm_mode=rank)"
float default="-1"
option "cutoff_value" U "The cutoff value (for --norm_mode=subtract_z)"
float default="-1.0"
option "exp" E "Raise the z-score to the power of this value (for --norm_mode=subtract_z)"
float default="-1.0"
section "Input"
option "input" i "Gene mapping file"
string typestr="filename" yes
section "Output"
option "dir_out" D "Output directory"
string typestr="directory" yes
