Evaluation metrics for a rank-list given some judgment gene-set. More...
#include <seekevaluate.h>
Static Public Member Functions | |
| static bool | SortRankVector (const vector< utype > &rank, const CSeekIntIntMap &mapG, vector< AResult > &a, const utype top=0) |
| Sort a gene ranking by the gene score. | |
| static bool | RankBiasedPrecision (const float &rate, const vector< utype > &rank, float &rbp, const vector< char > &mask, const vector< char > &gold, const CSeekIntIntMap &mapG, vector< AResult > *sing, const utype top=0) |
| Calculate the rank-biased precision for a gene ranking. | |
| static bool | AveragePrecision (const vector< utype > &rank, float &ap, const vector< char > &mask, const vector< char > &gold, const CSeekIntIntMap &mapG, vector< AResult > *ar) |
| Calculate the average precision for a gene ranking. | |
Detailed Description
Evaluation metrics for a rank-list given some judgment gene-set.
Provide static utility functions for evaluating a ranking of genes with the user-given gold standard gene-set. The typical use of such functions is in weighting datasets. Generally speaking, each dataset is weighted by how well the query genes are able to retrieve each other in the dataset. It is important to pick an informative measure to evaluate the retrieval of the query genes. Seek provides the choice of two evaluation metrics: Rank-Biased Precision (RBP) or Average Precision.
Definition at line 96 of file seekevaluate.h.
Member Function Documentation
| bool Sleipnir::CSeekPerformanceMeasure::AveragePrecision | ( | const vector< utype > & | rank, |
| float & | ap, | ||
| const vector< char > & | mask, | ||
| const vector< char > & | gold, | ||
| const CSeekIntIntMap & | mapG, | ||
| vector< AResult > * | ar | ||
| ) | [static] |
Calculate the average precision for a gene ranking.
- Parameters:
-
rank The gene-score vector ap The calculated average precision mask The genes in the ranking to be skipped over (typically the query genes) gold The gold-standard genes mapG The gene presence map. Genes that are not present in the dataset are skipped over. ar The sorted vector of (gene ID, gene score) pairs
Definition at line 111 of file seekevaluate.cpp.
References SortRankVector().
| bool Sleipnir::CSeekPerformanceMeasure::RankBiasedPrecision | ( | const float & | rate, |
| const vector< utype > & | rank, | ||
| float & | rbp, | ||
| const vector< char > & | mask, | ||
| const vector< char > & | gold, | ||
| const CSeekIntIntMap & | mapG, | ||
| vector< AResult > * | sing, | ||
| const utype | top = 0 |
||
| ) | [static] |
Calculate the rank-biased precision for a gene ranking.
- Parameters:
-
rate The parameter p in the RBP formula rbp The calculated RBP score, the output mask The genes in the ranking to be skipped over (typically the query genes) gold The gold-standard genes mapG The gene presence map. Genes that are not present in the dataset are skipped over. sing The sorted vector of (gene ID, gene score) pairs rank The gene-score vector top If X, sort only the topXelements. If 0, then sort the entire vector.
First calls the CSeekPerformanceMeasure::SortRankVector() with the arguments rank and top, in order to sort the gene-scores. Then with the sorted gene-ranking returned to sing, it calculates the rank-biased precision.
- Remarks:
- The RBP formula is given by:
where![\[RBP=\sum_{g \in U}{(1-p)p^{rank(g)}}\]](form_28.png)
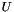 is the gold standard gene-set,
is the gold standard gene-set, 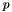 is the emphasis on ranks,
is the emphasis on ranks, 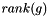 is the position of
is the position of 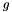 in the ranking is typically set to 0.95 - 0.99. The recommended value is 0.99. For more information, please read (Moffat et al 2008).
in the ranking is typically set to 0.95 - 0.99. The recommended value is 0.99. For more information, please read (Moffat et al 2008).
Definition at line 67 of file seekevaluate.cpp.
References SortRankVector().
Referenced by Sleipnir::CSeekWeighter::CVWeighting(), and Sleipnir::CSeekWeighter::OneGeneWeighting().
| bool Sleipnir::CSeekPerformanceMeasure::SortRankVector | ( | const vector< utype > & | rank, |
| const CSeekIntIntMap & | mapG, | ||
| vector< AResult > & | a, | ||
| const utype | top = 0 |
||
| ) | [static] |
Sort a gene ranking by the gene score.
- Parameters:
-
rank The vector of gene-scores to be sorted. Gene scores are inserted to this vector based on their gene IDs, which are a value from 0 to the size of the vector. mapG The gene presence map a The output, which is a vector of (gene ID, gene score) pairs that are sorted by score top If X, sort only the topXelements. If 0, then sort the entire vector.
The struct AResult represents a (gene ID, gene score) pair. This function sorts the vector of AResult in the descending order of the gene score.
Definition at line 25 of file seekevaluate.cpp.
References Sleipnir::CSeekIntIntMap::GetNumSet().
Referenced by AveragePrecision(), and RankBiasedPrecision().
The documentation for this class was generated from the following files:
- src/seekevaluate.h
- src/seekevaluate.cpp
