A suite of search algorithms that are supported by Seek. More...
#include <seekcentral.h>
Public Types | |
| enum | SearchMode { CV = 0, EQUAL = 1, USE_WEIGHT = 2, CV_CUSTOM = 3, ORDER_STATISTICS = 4, AVERAGE_Z = 5 } |
| Search modes (see section Detailed Descriptions) More... | |
Public Member Functions | |
| CSeekCentral () | |
| Constructor. | |
| ~CSeekCentral () | |
| Destructor. | |
| bool | Initialize (const vector< CSeekDBSetting * > &vecDBSetting, const char *search_dset, const char *query, const char *output_dir, const utype buffer=20, const bool to_output_text=false, const bool bOutputWeightComponent=false, const bool bSimulateWeight=false, const enum CSeekDataset::DistanceMeasure dist_measure=CSeekDataset::Z_SCORE, const bool bSubtractAvg=true, const bool bNormPlatform=false, const bool bLogit=false, const float fCutOff=-9999, const float fPercentQueryRequired=0, const float fPercentGenomeRequired=0, const bool bSquareZ=false, const bool bRandom=false, const int iNumRandom=10, gsl_rng *rand=NULL, const bool useNibble=false, const int numThreads=8) |
| Initialize function. | |
| bool | Initialize (const vector< CSeekDBSetting * > &vecDBSetting, const utype buffer=20, const bool to_output_text=false, const bool bOutputWeightComponent=false, const bool bSimulateWeight=false, const enum CSeekDataset::DistanceMeasure dist_measure=CSeekDataset::Z_SCORE, const bool bSubtractAvg=true, const bool bNormPlatform=false, const bool bLogit=false, const float fCutOff=-9999, const float fPercentQueryRequired=0, const float fPercentGenomeRequired=0, const bool bSquareZ=false, const bool bRandom=false, const int iNumRandom=10, gsl_rng *rand=NULL, const bool useNibble=false, const int numThreads=8) |
| Initialize function. | |
| bool | Initialize (const string &output_dir, const string &query, const string &search_dset, CSeekCentral *src, const int iClient, const float query_min_required=0, const float genome_min_required=0, const enum CSeekDataset::DistanceMeasure=CSeekDataset::Z_SCORE, const bool bSubtractGeneAvg=true, const bool bNormPlatform=false) |
| Initialize function. | |
| bool | CVSearch (gsl_rng *, const CSeekQuery::PartitionMode &, const utype &, const float &) |
| Run Seek with the cross-validated dataset weighting. | |
| bool | CVCustomSearch (const vector< vector< string > > &, gsl_rng *, const CSeekQuery::PartitionMode &, const utype &, const float &) |
| Run Seek with the custom dataset weighting. | |
| bool | EqualWeightSearch () |
| Run Seek with the equal dataset weighting. | |
| bool | WeightSearch (const vector< vector< float > > &) |
| Run Seek with the user-given dataset weights. | |
| bool | VarianceWeightSearch () |
| Run Seek with the variance weighted search. | |
| bool | AverageWeightSearch () |
| Run Seek with the SPELL search. | |
| bool | OrderStatistics () |
| Run Seek with the order statistics dataset weighting algorithm. | |
| const vector< vector < AResultFloat > > & | GetAllResult () const |
| Get the final gene-ranking for all the queries. | |
| const vector< CSeekQuery > & | GetAllQuery () const |
| Get all the queries. | |
| const vector< vector< float > > & | GetAllWeight () const |
| Get the dataset weight vector for all the queries. | |
| utype | GetGene (const string &strGene) const |
| Get the gene-map ID for a given gene-name. | |
| string | GetGene (const utype &geneID) const |
| Get the gene-name for a given gene-map ID. | |
| bool | Destruct () |
| Destruct this search instance. | |
| int | GetMaxGenomeCoverage () |
| Get the maximum genome coverage among the datasets in the compendium. | |
Detailed Description
A suite of search algorithms that are supported by Seek.
The Seek search algorithms perform the coexpression search of the user's query genes in a large compendium of microarray datasets. The output of the search algorithms is a ranking of genes based on their gene score, which is determined by the overall weighted coexpression to the query genes.
One of the first steps in a search is to weight the datasets in such a way to prioritize informative datasets. Then, with the weights generated, the final gene-score is given by:
![\[FS(g, Q)=\alpha\sum_{d \in D}{w_d \cdot s_d(g, Q)}\]](form_20.png)
where 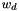 is the weight of the dataset,
is the weight of the dataset, 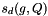 is the score of
is the score of 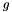 to the query in the dataset,
to the query in the dataset, 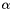 is the normalization constant.
is the normalization constant.
Currently the following dataset weighting algorithms are supported in Seek.
- The query cross-validated (CV) weighting (CSeekCentral::CV): This is a weighting based on the query coexpression. The idea is to measure how well query genes are able to retrieve each other under a cross-validation setting. To do so, we first divide the query into N parts, use 1 part to build a small search instance, and use
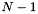 parts for evaluating the instance. The score of each instance
parts for evaluating the instance. The score of each instance 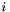 is given by:
is given by:
where![\[s(i)=\sum_{g \in U}{(1-p)p^{rank(g)}}\]](form_23.png)
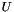 is the genes in parts,
is the genes in parts, 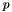 is an exponential rate parameter,
is an exponential rate parameter, 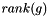 is the position of in the ranking of genes generated by the search instance.
is the position of in the ranking of genes generated by the search instance.
- Equal weighting (CSeekCentral::EQUAL): the weight is 1 for all datasets.
- User-supplied weight vector (CSeekCentral::USE_WEIGHT). (ie., Seek does not calculate dataset weights)
- User-supplied gene-sets for weighting datasets, and also use cross-validations (CSeekCentral::CV_CUSTOM)
- Order-statistics (CSeekCentral::ORDER_STATISTICS): the algorithm used in MEM. (Adler et al, Genome Biology 2009)
CSeekCentral can handle multiple queries at a time, but the search parameters must remain the same for all queries.
Definition at line 81 of file seekcentral.h.
Member Enumeration Documentation
Search modes (see section Detailed Descriptions)
- Enumerator:
Definition at line 88 of file seekcentral.h.
Member Function Documentation
Run Seek with the SPELL search.
- Remarks:
- Assumes that the CSeekCentral::Initialize() has been called.
Definition at line 1431 of file seekcentral.cpp.
References AVERAGE_Z.
| bool Sleipnir::CSeekCentral::CVCustomSearch | ( | const vector< vector< string > > & | newGoldStd, |
| gsl_rng * | rnd, | ||
| const CSeekQuery::PartitionMode & | PART_M, | ||
| const utype & | FOLD, | ||
| const float & | RATE | ||
| ) |
Run Seek with the custom dataset weighting.
- Parameters:
-
newGoldStd The gold-standard gene-set that is used for weighting datasets rnd The random number generator PART_M Query partition mode FOLD Number of partitions to generate from the query RATE The weighting parameter p * Same as CVSearch, except that the weighting is not based on the coexpression of the query genes, but based on the similarity of the query genes to some custom gold standard gene-set.
- Remarks:
- The random number generator is used for partitioning the query.
- Assumes that the CSeekCentral::Initialize() has been called.
Definition at line 1413 of file seekcentral.cpp.
References CV_CUSTOM.
| bool Sleipnir::CSeekCentral::CVSearch | ( | gsl_rng * | rnd, |
| const CSeekQuery::PartitionMode & | PART_M, | ||
| const utype & | FOLD, | ||
| const float & | RATE | ||
| ) |
Run Seek with the cross-validated dataset weighting.
- Parameters:
-
rnd The random number generator PART_M Query partition mode FOLD Number of partitions to generate from the query RATE The weighting parameter p
- Remarks:
- The random number generator is used for partitioning the query.
- Assumes that the CSeekCentral::Initialize() has been called.
Definition at line 1404 of file seekcentral.cpp.
References CV.
| bool Sleipnir::CSeekCentral::Destruct | ( | ) |
Destruct this search instance.
- Returns:
- True if successful.
Definition at line 1463 of file seekcentral.cpp.
Run Seek with the equal dataset weighting.
- Remarks:
- Assumes that the CSeekCentral::Initialize() has been called.
Definition at line 1399 of file seekcentral.cpp.
References EQUAL.
| const vector< CSeekQuery > & Sleipnir::CSeekCentral::GetAllQuery | ( | ) | const |
| const vector< vector< AResultFloat > > & Sleipnir::CSeekCentral::GetAllResult | ( | ) | const |
Get the final gene-ranking for all the queries.
- Returns:
- A two-dimensional array that stores the gene-rankings
Definition at line 1474 of file seekcentral.cpp.
| const vector< vector< float > > & Sleipnir::CSeekCentral::GetAllWeight | ( | ) | const |
Get the dataset weight vector for all the queries.
- Returns:
- A two-dimensional
floatarray that stores the weights
- Remarks:
- The first dimension is the query. The second dimension is the dataset.
Definition at line 1491 of file seekcentral.cpp.
| utype Sleipnir::CSeekCentral::GetGene | ( | const string & | strGene | ) | const |
Get the gene-map ID for a given gene-name.
- Parameters:
-
strGene The gene-name as a string
- Returns:
- The gene-map ID
Definition at line 1482 of file seekcentral.cpp.
References Sleipnir::CSeekTools::GetNaN().
| string Sleipnir::CSeekCentral::GetGene | ( | const utype & | geneID | ) | const |
Get the gene-name for a given gene-map ID.
- Parameters:
-
geneID The gene-map ID
- Returns:
- The gene-name as a
string
Definition at line 1487 of file seekcentral.cpp.
| bool Sleipnir::CSeekCentral::Initialize | ( | const vector< CSeekDBSetting * > & | vecDBSetting, |
| const char * | search_dset, | ||
| const char * | query, | ||
| const char * | output_dir, | ||
| const utype | buffer = 20, |
||
| const bool | to_output_text = false, |
||
| const bool | bOutputWeightComponent = false, |
||
| const bool | bSimulateWeight = false, |
||
| const enum CSeekDataset::DistanceMeasure | dist_measure = CSeekDataset::Z_SCORE, |
||
| const bool | bSubtractAvg = true, |
||
| const bool | bNormPlatform = false, |
||
| const bool | bLogit = false, |
||
| const float | fCutOff = -9999, |
||
| const float | fPercentQueryRequired = 0, |
||
| const float | fPercentGenomeRequired = 0, |
||
| const bool | bSquareZ = false, |
||
| const bool | bRandom = false, |
||
| const int | iNumRandom = 10, |
||
| gsl_rng * | rand = NULL, |
||
| const bool | useNibble = false, |
||
| const int | numThreads = 8 |
||
| ) |
Initialize function.
Performs the following operations:
- Read the search parameters
- Read the gene mapping
gene_map.txt - Read a list of queries
- Read the dataset mapping and the search datasets
- Read the CDatabaselets (ie, the gene-gene correlations for the query genes)
- Parameters:
-
gene The gene mapping file name, gene_map.txtquant The quant file name dset The dataset mapping file name, dataset_platform.txtsearch_dset The file which contains the dataset names to be used for the search query The query file name platform The platform directory, which contains the platform correlation averages and standard deviations db The CDatabaselet directory, which contains the gene-centric compendium-wide correlations, *.db filesprep The Prep directory, which contains the gene correlation average *.gavg, and the gene presence*.gpres.gvar The gene variance directory, which contains the *.gvar filessinfo The sinfo directory, which contains the *.sinfo filesnum_db The total number of CDatabaselet files buffer The number of query genes to store in the memory output_dir The output directory to_output_text If true, output the gene-ranking in textual format bOutputWeightComponent If true, output the dataset weight components (ie the score of cross-validations) bSimulateWeight If true, use simulated weight as dataset weight dist_measure Distance measure, either CORRELATION or Z_SCORE bSubtractAvg If true, subtract the average z-score on a per-gene basis bNormPlatform If true, subtract the platform gene average, divide by platform gene standard deviation bLogit If true, apply the logit transformation on the correlations fCutOff Cutoff the correlation values fPercentRequired The fraction of the query genes required to be present in a dataset in order to consider the dataset for integration bSquareZ If true, square the correlations bRandom If true, shuffle the correlation vector iNumRandom The number of random simulations to perform per query rand The random number generator useNibble Default to false
- Remarks:
- The word correlation refers to the z-scored, standardized Pearson.
-
The parameters
bSubtractAvg,bNormPlatform,bLogit, andbSquareZare options to transform the correlation values. -
The
bSimulateWeightoption is for equal weighting or order statistics where the final gene ranking is not derived from a weighted integration of datasets. In this case, if the user still wants to see the contribution of each dataset, the simulated weight is computed from the distance of a dataset's coexpression ranking to the final gene ranking. - This function is designed to be used by SeekMiner.
Definition at line 655 of file seekcentral.cpp.
References Sleipnir::CSeekTools::ReadMultipleQueries().
| bool Sleipnir::CSeekCentral::Initialize | ( | const vector< CSeekDBSetting * > & | vecDBSetting, |
| const utype | buffer = 20, |
||
| const bool | to_output_text = false, |
||
| const bool | bOutputWeightComponent = false, |
||
| const bool | bSimulateWeight = false, |
||
| const enum CSeekDataset::DistanceMeasure | dist_measure = CSeekDataset::Z_SCORE, |
||
| const bool | bSubtractAvg = true, |
||
| const bool | bNormPlatform = false, |
||
| const bool | bLogit = false, |
||
| const float | fCutOff = -9999, |
||
| const float | fPercentQueryRequired = 0, |
||
| const float | fPercentGenomeRequired = 0, |
||
| const bool | bSquareZ = false, |
||
| const bool | bRandom = false, |
||
| const int | iNumRandom = 10, |
||
| gsl_rng * | rand = NULL, |
||
| const bool | useNibble = false, |
||
| const int | numThreads = 8 |
||
| ) |
Initialize function.
Load everything except the query, the search datasets, and the output directory
- Parameters:
-
gene The gene mapping file name, gene_map.txtquant The quant file name dset The dataset mapping file name, dataset_platform.txtplatform The platform directory, which contains the platform correlation average and standard deviation db The CDatabaselet directory, which contains the gene-centric compendium-wide correlations, *.db filesprep The Prep directory, which contains the gene correlation average *.gavg, and the gene presence*.gpres. Divided by datasets.gvar The gene variance directory, which contains the *.gvar filessinfo The sinfo directory, which contains the *.sinfo filesnum_db The total number of CDatabaselet files buffer The number of query genes to store in the memory to_output_text If true, output the gene-ranking in the textual format bOutputWeightComponent If true, output the dataset weight components (ie the score of cross-validations) bSimulateWeight If true, use simulated weight as dataset weight dist_measure Distance measure, either CORRELATION or Z_SCORE bSubtractAvg If true, subtract the average z-score on a per-gene basis bNormPlatform If true, subtract the platform gene average, divide by platform gene standard deviation bLogit If true, apply the logit transformation on the correlations fCutOff Cutoff the correlations fPercentRequired The fraction of the query genes required to be present in a dataset bSquareZ If true, square the correlations bRandom If true, shuffle the correlation vector iNumRandom The number of random simulations to perform per query rand The random number generator useNibble Default to false
- Remarks:
- The word correlation refers to the z-scored, standardized Pearson.
-
The parameters
bSubtractAvg,bNormPlatform,bLogit, andbSquareZare options to transform the correlation values. -
The
bSimulateWeightoption is for equal weighting or order statistics where the final gene ranking is not derived from a weighted integration of datasets. In this case, if the user still wants to see the contribution of each dataset, the simulated weight is computed from the distance of a dataset's coexpression ranking to the final gene ranking. - This function is designed to be used by SeekMiner.
Definition at line 521 of file seekcentral.cpp.
References Sleipnir::CSeekDataset::CORRELATION, Sleipnir::CSeekTools::LoadDatabase(), Sleipnir::CSeekTools::ReadListTwoColumns(), Sleipnir::CSeekTools::ReadPlatforms(), and Sleipnir::CSeekTools::ReadQuantFile().
| bool Sleipnir::CSeekCentral::Initialize | ( | const string & | output_dir, |
| const string & | query, | ||
| const string & | search_dset, | ||
| CSeekCentral * | src, | ||
| const int | iClient, | ||
| const float | query_min_required = 0, |
||
| const float | genome_min_required = 0, |
||
| const enum CSeekDataset::DistanceMeasure | eDistMeasure = CSeekDataset::Z_SCORE, |
||
| const bool | bSubtractGeneAvg = true, |
||
| const bool | bNormPlatform = false |
||
| ) |
Initialize function.
Prepares Seek to be used in a client-server environment
- Parameters:
-
output_dir The output directory query The query file name search_dset The file that contains the name of datasets to be used for the search src The CSeekCentral instance, where some settings will be copied to here iClient The client's socket connection query_min_required The minimum number of query genes required to be present in a dataset dist_measure Distance measure, either CORRELATION or Z_SCORE. bSubtractAvg If true, subtract the average z-score on a per-gene basis bNormPlatform If true, subtract the platform gene average, divide by platform gene standard deviation
- Remarks:
- This function is designed to be used by SeekServer.
-
The parameters
bSubtractAvg,bNormPlatformare options to transform the correlation values. -
Assumes that the CDatabaselets have been read, and the
*.gvar,*.sinfo files have been loaded. - Assumes that the dataset and gene mapping files have been read.
Definition at line 237 of file seekcentral.cpp.
References Sleipnir::CSeekTools::LoadDatabase(), and Sleipnir::CMeta::Tokenize().
Run Seek with the order statistics dataset weighting algorithm.
- Remarks:
- Assumes that the CSeekCentral::Initialize() has been called.
Definition at line 1426 of file seekcentral.cpp.
References ORDER_STATISTICS.
Run Seek with the variance weighted search.
Same as CSeekCentral::WeightSearch(), except that the user-given weights are the query gene expression variances.
- Remarks:
- Assumes that the CSeekCentral::Initialize() has been called.
Definition at line 1436 of file seekcentral.cpp.
References Sleipnir::CSeekQuery::GetQuery(), Sleipnir::CSeekTools::InitVector(), Sleipnir::CMeta::IsNaN(), and WeightSearch().
| bool Sleipnir::CSeekCentral::WeightSearch | ( | const vector< vector< float > > & | weights | ) |
Run Seek with the user-given dataset weights.
- Parameters:
-
weights A two-dimensional array that stores the user-given weights
- Remarks:
- The two-dimensional array
weightsis Q by D : where Q is the number of queries, D is the number of datasets.weights[i][j] stores the weight of dataset j in query i. - Assumes that the CSeekCentral::Initialize() has been called.
Definition at line 1421 of file seekcentral.cpp.
References USE_WEIGHT.
Referenced by VarianceWeightSearch().
The documentation for this class was generated from the following files:
- src/seekcentral.h
- src/seekcentral.cpp
