Provide functions to assign dataset weight using the query gene. More...
#include <seekweight.h>
Static Public Member Functions | |
| static bool | LinearCombine (vector< utype > &rank, const vector< utype > &cv_query, CSeekDataset &sDataset, const utype &, const bool &) |
| Calculates for each gene the average correlation to all of the query genes in a dataset. | |
| static bool | CVWeighting (CSeekQuery &sQuery, CSeekDataset &sDataset, const float &rate, const float &percent_required, const bool &bsquareZ, vector< utype > *rrank, const CSeekQuery *goldStd=NULL) |
| Cross-validates query-genes in a dataset. | |
| static bool | OrderStatisticsRankAggregation (const utype &, const utype &, utype **, const vector< utype > &, vector< float > &, const utype &) |
| Performs OrderStatisticsAggregation, also known as the MEM algorithm. | |
| static bool | OrderStatisticsPreCompute () |
| static bool | OneGeneWeighting (CSeekQuery &, CSeekDataset &, const float &, const float &, const bool &, vector< utype > *, const CSeekQuery *) |
| Simulates a dataset weight for one-gene query. | |
| static bool | AverageWeighting (CSeekQuery &sQuery, CSeekDataset &sDataset, const float &percent_required, const bool &bSquareZ, float &w) |
Detailed Description
Provide functions to assign dataset weight using the query gene.
For dataset weighting, one way is to use CSeekWeighter::CVWeighting. The CSeekWeighter::CVWeighting uses a cross-validation (CV) framework, where it partitions the query and performs a search instance on one sub-query, using the remainder of the queries as the evaluation of the search instance.
The CSeekWeighter::OrderStatisticsRankAggregation is a rank-based technique described by Adler et al (2009). This combines dataset weighting and dataset gene-ranking aggregation all into one step.
Definition at line 44 of file seekweight.h.
Member Function Documentation
| bool Sleipnir::CSeekWeighter::CVWeighting | ( | CSeekQuery & | sQuery, |
| CSeekDataset & | sDataset, | ||
| const float & | rate, | ||
| const float & | percent_required, | ||
| const bool & | bsquareZ, | ||
| vector< utype > * | rrank, | ||
| const CSeekQuery * | goldStd = NULL |
||
| ) | [static] |
Cross-validates query-genes in a dataset.
- Parameters:
-
sQuery The query and its partitions sDataset A dataset rate RBP parameter p percent_required Percentage of query genes required to be present in the dataset bSquareZ Whether or not to square correlations rrank Temporary vector storing intermediary correlations goldStd If a gold-standard gene-set is provided, use this to evaluate the retrieval of a cross-validation
This performs multiple cross-validation runs to validate the query genes in retrieving themselves in the dataset. The sum of the evaluation of all the runs then becomes the dataset weight. For evaluation, we use the following formula for scoring a validation run 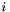 :
:
![\[s(i)=\sum_{g \in U}{(1-p)p^{rank(g)}}\]](form_23.png)
where 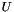 is the
is the 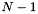 parts of the query used for evaluation,
parts of the query used for evaluation, 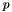 is an exponential rate parameter,
is an exponential rate parameter, 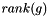 is the position of
is the position of 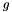 in the ranking of genes generated by the subsearch instance .
in the ranking of genes generated by the subsearch instance .
The above formulation is inspired by rank-biased precision. The parameter p needs to be provided. The default value is 0.99.
Definition at line 452 of file seekweight.cpp.
References Sleipnir::CSeekQuery::GetCVQuery(), Sleipnir::CSeekIntIntMap::GetForward(), Sleipnir::CSeekDataset::GetGeneMap(), Sleipnir::CSeekQuery::GetNumFold(), Sleipnir::CSeekDataset::GetNumGenes(), Sleipnir::CSeekQuery::GetQuery(), Sleipnir::CSeekDataset::GetQueryMap(), Sleipnir::CSeekDataset::InitializeCVWeight(), Sleipnir::CSeekTools::InitVector(), Sleipnir::CSeekTools::IsNaN(), LinearCombine(), Sleipnir::CSeekPerformanceMeasure::RankBiasedPrecision(), and Sleipnir::CSeekDataset::SetCVWeight().
| bool Sleipnir::CSeekWeighter::LinearCombine | ( | vector< utype > & | rank, |
| const vector< utype > & | cv_query, | ||
| CSeekDataset & | sDataset, | ||
| const utype & | MIN_REQUIRED, | ||
| const bool & | bSquareZ | ||
| ) | [static] |
Calculates for each gene the average correlation to all of the query genes in a dataset.
- Parameters:
-
rank A vector that stores the correlation of each gene to all of the query genes cv_query A vector that stores the query genes sDataset A dataset MIN_REQUIRED A utype that specifies how many query genes are required to be present in a dataset. If not enough query genes are present, then the averaging is not performed. bSquareZ If true, square the correlation values before adding correlations.
- Remarks:
- The word correlations refer to z-scored, standardized Pearson correlations. The result is returned in the parameter
rank.
Definition at line 30 of file seekweight.cpp.
References Sleipnir::CSeekDataset::GetDataMatrix(), Sleipnir::CSeekIntIntMap::GetForward(), Sleipnir::CSeekDataset::GetGeneMap(), Sleipnir::CSeekDataset::GetNumGenes(), Sleipnir::CSeekDataset::GetQueryMap(), and Sleipnir::CSeekTools::InitVector().
Referenced by CVWeighting(), and OneGeneWeighting().
| bool Sleipnir::CSeekWeighter::OneGeneWeighting | ( | CSeekQuery & | sQuery, |
| CSeekDataset & | sDataset, | ||
| const float & | rate, | ||
| const float & | percent_required, | ||
| const bool & | bSquareZ, | ||
| vector< utype > * | rrank, | ||
| const CSeekQuery * | goldStd | ||
| ) | [static] |
Simulates a dataset weight for one-gene query.
- Parameters:
-
sQuery The query sDataset The dataset rate RBP parameter p percent_required Percentage of query genes required to be present in a dataset (assumed to be 1 in this case) bSquareZ Whether or not to square correlations rrank Final gene-score goldStd Gold-standard gene-set for weighting a dataset
This function is mainly used for equal weighting. Although equal weighting integrates all datasets with weight = 1, for the purpose of displaying datasets, the datasets need to be ranked according to the distance to the average gene-ranking.
This average gene-ranking is produced by summing gene-rankings from all datasets and divided by the number of datasets. To score a dataset, we calculate the RBP precision of this dataset in retrieving the top 100 genes of the average ranking.
Definition at line 321 of file seekweight.cpp.
References Sleipnir::CSeekQuery::GetCVQuery(), Sleipnir::CSeekIntIntMap::GetForward(), Sleipnir::CSeekDataset::GetGeneMap(), Sleipnir::CSeekDataset::GetNumGenes(), Sleipnir::CSeekQuery::GetQuery(), Sleipnir::CSeekDataset::GetQueryMap(), Sleipnir::CSeekDataset::InitializeCVWeight(), Sleipnir::CSeekTools::InitVector(), Sleipnir::CSeekTools::IsNaN(), LinearCombine(), Sleipnir::CSeekPerformanceMeasure::RankBiasedPrecision(), and Sleipnir::CSeekDataset::SetCVWeight().
| bool Sleipnir::CSeekWeighter::OrderStatisticsRankAggregation | ( | const utype & | iDatasets, |
| const utype & | iGenes, | ||
| utype ** | rank_d, | ||
| const vector< utype > & | counts, | ||
| vector< float > & | master_rank, | ||
| const utype & | numThreads | ||
| ) | [static] |
Performs OrderStatisticsAggregation, also known as the MEM algorithm.
- Parameters:
-
iDatasets The number of datasets iGenes The number of genes rank_d Two-dimensional vectors storing correlation-ranks to the query genes. First dimension: datasets. Second dimension: genes. counts A vector storing the count of datasets for each gene master_rank A vector storing the integrated gene-score numThreads The number of threads to be used (in a parallel setup)
rank_d needs to be prepared as follows: a correlation rank vector is obtained from sorting Pearson correlations in a dataset, and then it is normalized by (rank of correlation) / (number of genes). The result is stored in rank_d.
Afterward, for each gene g, the algorithm compares this gene's rank_d distribution across datasets with that derived from a set of datasets with randomly ordered correlation vectors (ie a null distribution). A significance p-value is calculated for this gene, and -log(p) values are stored in master_rank.
Definition at line 175 of file seekweight.cpp.
References Sleipnir::CSeekTools::Free2DArray(), and Sleipnir::CSeekTools::Init2DArray().
The documentation for this class was generated from the following files:
- src/seekweight.h
- src/seekweight.cpp
